Méthylation de l’ADN et ARN Natifs
Plusieurs méthodes d’analyse du degré de méthylation de l’ADN ont été mises au point pour déterminer statut hypo ou hyperméthylé d’un grand nombre d’ilots CpG, voire du génome entier. La plus couramment utilisée est basée sur la conversion au bisulfite des cytosines non méthylées en uracile avec, cependant, plusieurs inconvénients dont la complexité de la préparation de la librairie, les biais de conversion et l’impossibilité de distinguer entre les modifications 5mC et 5hmC.
L’ARN messager peut aussi porter des modifications (épitranscriptome) dont la plus fréquente chez les eucaryotes est la N6-Methyladenosine (m6A) qui influence l’expression des protéines, en régulant par exemple l’épissage ou la stabilité des ARNs . Le niveau de m6A est généralement quantifié par immunoprecipation avec des difficultés de spécificité et de rendement (Deng X. et al. 2023).
Pour une vision plus précise de la complexité du génome et du transcriptome, les technologies de séquençage sur longs fragments (PacBio Hifi, Nanopore ONT) ont permis une avancée dans l’analyse de l’ADN et de l’ARN natif.
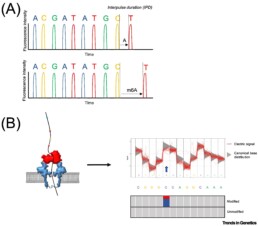
Avec ces techniques, les modifications 5mC et 6mC de l’ADN ou de l’ARN (sans traitement préalable altérant l’échantillon) sont détectées par un changement de signal (durée d’interpulsion (IPD) ou différence de tension). L’état de méthylation se mesure par les écarts à la distributions canonique qui induit des erreurs de base-calling, localisées, selon le type de modification, au niveau des nucléotides méthylés ou des positions adjacentes. (voir figure issue de van Dijk E.L. et al. 2023)
L’analyse reste une étape délicate. Les logiciels utilisés varient selon les approches de détection choisies, ceux qui utilisent les profils de séquençage ou bien les erreurs de « base calling ». Depuis 2017, de nombreux développement ont été réalisé sur les outils bioinformatiques comme décrit par Furlan M. et al. 2021.
Rubriques associées
- Séquençage des petits ARN
- Cartographie des sites d’initiation de la transcription : TSS seq
- TAPS/TAPSβ
- Enzymatic Methyl-seq (EM-seq™)
- Cartographie des sites ADN – Protéines de la chromatine : CUT & RUN vs CUT & Tag
- Cartographie des contacts de la chromatine : 3C, 4C et 5C
- Cartographie des contacts cis et trans de la chromatine : HiC-seq
- Cartographie des sites d’accessibilité de la chromatine : DNase-seq
- Cartographie indirecte des sites d’accessibilité de la chromatine : MNase seq
- Cartographie des sites d’accessibilité de la chromatine : FAIRE seq
- Cartographie des sites d’accessibilité de la chromatine : ATAC seq
- Cartographie des sites des interactions ARN- protéines : CLIP seq
- Cartographie des sites des interactions ADN- protéines : CHIP seq
- Cartographie des marques épigénétiques de l’ADN : MeDIP
- Cartographie des marques épigénétiques de l’ADN : Methyl seq
- BiSeq