Omni-C
Protocole de ligation de proximité utilisant une endonucléase indépendante de la séquence, générant des données pour l’identification des TAD et le scaffolding [1].
Le kit Omni-C® de Dovetail® produit des librairies de ligature de proximité de haute qualité. Ce protocole augmente la couverture génomique et réduit les biais par rapport aux préparations Hi-C basées sur les enzymes de restriction. Ce protocole est utilisé pour l’identification des Domaines d’association topologique (TAD), ou lorsqu’une couverture homogène du génome est nécessaire, par exemple pour la détermination des SNP et le génotypage, c’est également une méthode de scaffolding efficace. Aucune extraction n’est nécessaire, car la préparation commence directement à partir de cellules ou de tissus finement broyés.
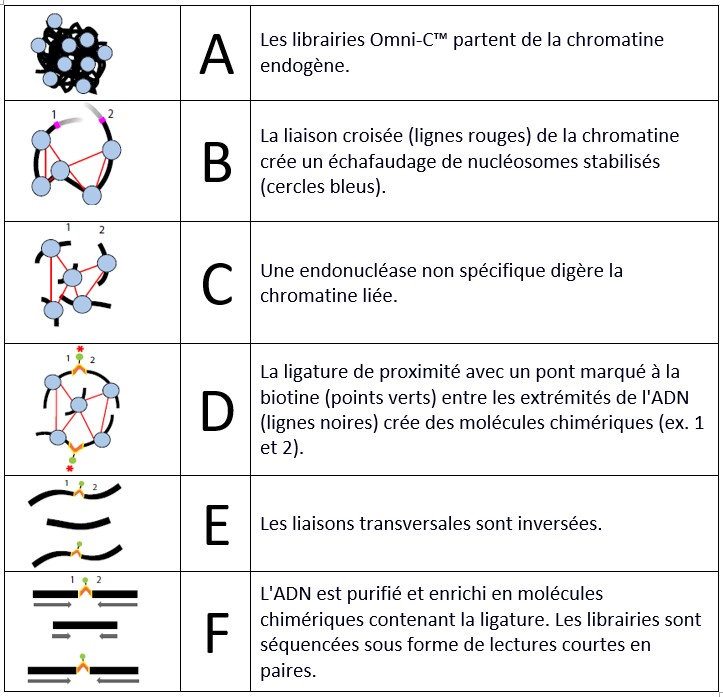
La préparation de la librairie se fait en 5 étapes :
- Préparation de l’échantillon et réticulation
La chromatine est fixée à l’aide de formaldéhyde. Après l’arrêt de la réticulation, une digestion in situ de la chromatine est effectuée à l’aide d’une enzyme endonucléase.
- Quantification du lysat
La chromatine est libérée par la lyse des cellules. Dans cette étape, la quantité de chromatine obtenue ainsi que le degré de digestion sont évalués pour assurer le succès de la préparation de la librairie.
- Ligation de proximité
Polissage des extrémités, ligature d’un pont d’oligonucléotides biotinylés. La ligature intra-agrégat pour capturer les contacts est effectuée, suivie de l’inversion de la réticulation et de la purification de l’ADN.
- Préparation de la librairie
Les étapes de réparation des extrémités, de ligature des adaptateurs et de purification permettent d’obtenir le modèle pour l’étape finale.
- Capture et amplification de la ligature
Une étape d’enrichissement à la streptavidine permet de capturer les produits de l’étape de ligature de proximité. Des index sont ajoutés par PCR et une purification sur billes ainsi qu’une sélection de taille permettent d’obtenir des librairies compatibles avec Illumina. Il est possible de multiplexer jusqu’à 24 librairies.
Le résultat attendu est des librairies séquencées contenant des informations sur les parties du génome qui sont physiquement proches dans les noyaux des cellules ou des tissus utilisés. Ces informations peuvent être utilisées pour établir des cartes de contact des échantillons après avoir aligné les lectures sur le génome de référence ou pour assembler un génome en scaffolds à l’échelle d’un chromosome ou d’un quasi-chromosome.
Quel que soit le type d’échantillon, Omni-C doit permettre de générer des librairies très complexes et contenant beaucoup d’information, même à faible concentration initiale. Par rapport à d’autres données Hi-C basées sur des enzymes de restriction, l’attendu est une couverture plus uniformément répartie sur le génome. Cela permet des analyses en aval telles que la détermination des SNP et le phasage.
La couverture uniforme des données Omni-C doit également rendre plus précis le scaffolding des contigs pour l’assemblage du génome, car aucun contig ne manquera en raison de l’absence de sites de restriction, comme cela peut se produire avec les données Hi-C classiques.
[1] https://ngisweden.scilifelab.se/methods/dovetail-omni-c/
[2] https://dovetailgenomics.com/wp-content/uploads/2021/09/Omni-C-Tech-Note.pdf