Le séquençage du génome entier
L’approche utilisée pour l’étude du génome d’un organisme diffère selon l’enjeu du séquençage.
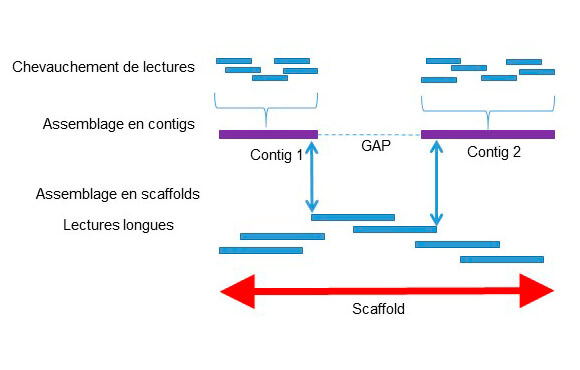
Le séquençage de novo permet de reconstituer un génome inconnu, non référencé dans les bases de données, grâce à l’assemblage de données de séquences. Les outils bioinformatiques utilisent le chevauchement des séquences pour construire des contigs de taille la plus longue possible. Les contig sont eux même assemblés les uns aux autres pour créer des « scaffolds » et générer ainsi un alignement de séquences sur toute la longueur du génome. Selon la technologie utilisée, il est nécessaire de combiner un séquençage à courtes et longues lectures pour plus de précision dans l’assemblage (Fig1).
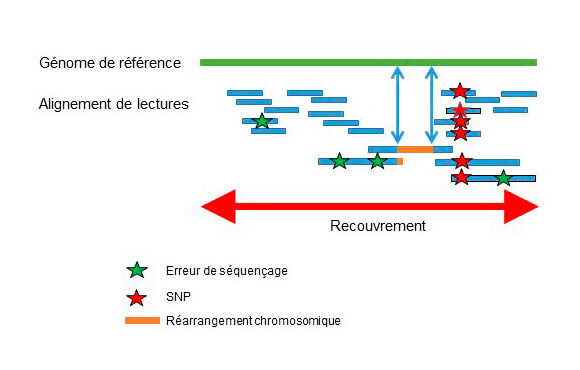
Le séquençage de génomes déjà connus (reséquençage ou WGS) a pour enjeu majeur de répertorier les variations nucléotidiques et structurales et de comprendre leurs conséquences biologiques. Les séquences courtes issues du reséquencage sont alignées sur la séquence de référence pour identifier les variations génomiques de type SNP. L’intégration de lectures longues à un séquençage à lectures courtes est recommandée pour la détection précise des CNV, indels et les réarrangements chromosomiques (Fig2).
Le séquençage « en paire » (paired end) consiste à séquencer les deux extrémités de fragments courts, de taille inférieure à 1kb (lectures courtes).
Plusieurs méthodologies sont disponibles pour produire des séquences dites à longues lectures :
– la production de banques « Mate Pair» permet de séquencer les deux extrémités de fragments de plusieurs kilobases (kb).
– la technologie « SMRT» de PacBio (RSII, Sequel) permet de produire un séquençage à longues lectures (>20kb), et d’obtenir, en théorie, un taux de couverture suffisant pour un assemblage de novo, mais son coût élevé rend difficile son emploi pour les grands génomes eucaryotes.
– la technologie des lectures longues synthétiques est basée sur un système d’indexage moléculaire de longs fragments (jusqu’à 100kb) permettant de reliées physiquement entre elles des séquences courtes issues de ces grands fragments. Cette stratégie est proposée par la technologie 10X Genomics (GemCode et Chromium) pour faciliter l’assemblage.
– la technologie nanopore de Oxford Nanopore Technology permet de produire un séquençage à très longues lectures (>2Mb), mais produit encore un taux d’erreur de lecture élevé qui peut être corrigé par association avec un séquençage à lectures courtes plus précis.
– la carte optique en complément du séquençage en paire permet de réaliser un assemblage précis en particulier pour les génomes microbiens.