Adaptive sampling
Lorsque des expériences de séquençage à grande échelle sont effectuées, il peut être difficile de suivre la région d’intérêt dans un ensemble de données en expansion rapide. L’isolement de l’ADN d’intérêt avant le séquençage peut nécessiter une préparation d’échantillons fastidieuse.
Oxford Nanopore Technology (ONT) a développé une méthode de séquençage à lecture longue connue sous le nom de séquençage « adaptatif ».
Grâce à l’échantillonnage adaptatif ou ‘adaptive sampling’ (AS), c’est le séquenceur qui sélectionne la région d’intérêt. Au cours du séquençage ONT typique, les brins d’ADN sont lus via des changements de potentiel électrique lorsque les molécules transitent à travers les pores d’une membrane spécialement préparée. Contrairement au séquençage Illumina, ces données de lecture ONT sont disponibles en temps réel pendant le run, plutôt que d’être disponibles uniquement lorsque le run est terminé.
Dans l’AS, les données de séquence sont à la fois générées et comparées en temps réel à celles d’une région d’intérêt sélectionnée dans le génome. Si le brin d’ADN entrant dans le pore ne présente pas d’intérêt, le flux de données en temps réel le signale (la tension est inversée) et le brin est éjecté du nanopore, laissant le pore libre pour le brin suivant de l’échantillon.
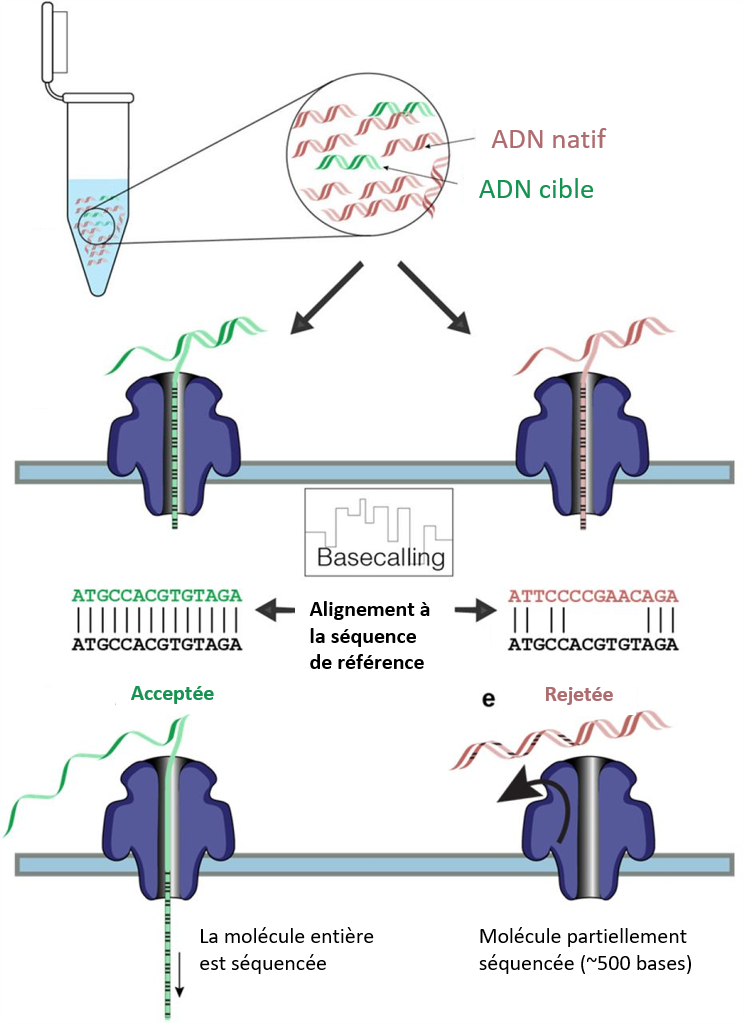
Au fur et à mesure que le brin d’intérêt est traité, le système peut rapidement voir qu’il s’agit d’un brin cible et le séquençage se poursuit. Cela se traduit par une couverture élevée des données de séquençage pour la région d’intérêt.
L’utilisateur fournit la séquence FASTA du génome de référence ainsi que les coordonnées de la région d’intérêt. Plusieurs régions d’intérêt différentes qui ne se chevauchent pas peuvent être échantillonnées simultanément.
Cette technologie permet l’enrichissement des échantillons sans les préparations de banques intensives et fastidieuses requises, par exemple, par l’enrichissement ciblé Cas9 pour le séquençage Nanopore. L’AS augmente la proportion de lectures de faible abondance dans les échantillons métagénomiques (Martin et al. 2022) et a le potentiel de caractériser des régions de forte variance (Weilguny et al. 2023).