ScNaUmi-seq
L’approche conventionnelle du scRNAseq est basée sur le séquençage de courts fragments de cDNAs indexés (voir scRNAseq). L’analyse des « cDNAs pleine longueur » nécessite, de ce fait, un traitement bioinformatique d’assemblage qui complique, par exemple, la détection d’isoformes de transcrits. Pour y palier, le séquençage en longue lecture par les technologies PacBio ou Nanopore (ONT) serait plus approprié.
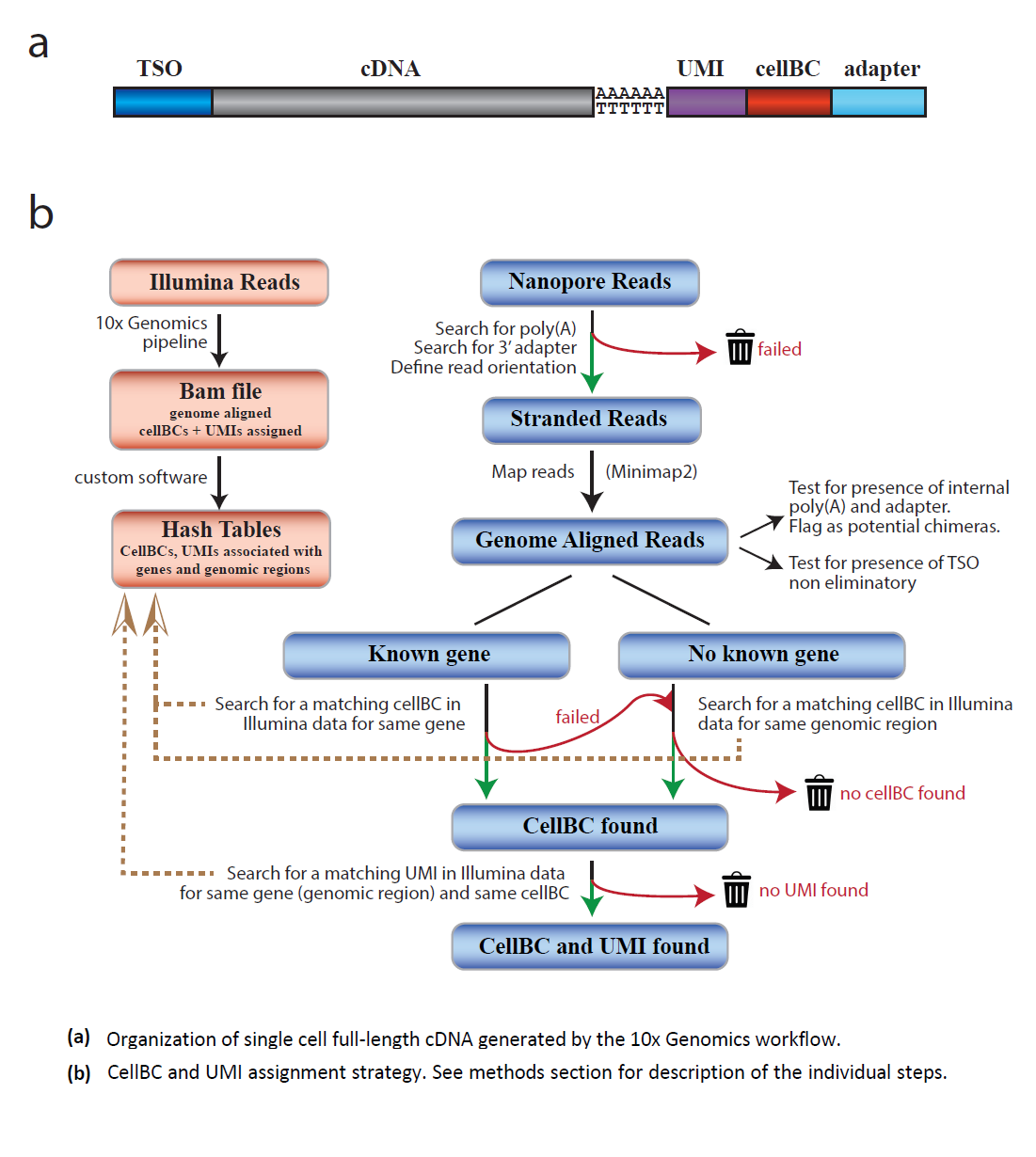
La méthode ScNaUmi-seq (Lebrigand et al. 2020) utilise la même stratégie que le Dropseq pour la capture et l’indexage en 3’des RNApoly A (Figure) suivi d’un séquençage Nanopore. Le traitement des données, basé sur l’assignation précise des cellBC et UMIs, suit plusieurs étapes (Figure) qui permettent de réduire les artefacts de RT ou de PCR et d’identifier de réelles chimères de transcrits et des isoformes rares. L’amélioration des kits de préparation des banques de cDNAs (Smart seq v4, kit ONT V14,etc…), de la chimie Nanopore et qualité d’analyse des lectures devrait faciliter l’étude du transcriptome cellulaire (cf Nanopore 2022)
Plateformes à contacter pour ce domaine d’expertise
Mise à jour : Avril 2024